Some of the language surrounding high-throughput sequencing can be hard to get to grips with. Importantly, this can lead to miscommunication, misunderstandings and misinterpretation of data. So this will quickly highlight some of the lingo used and some of the common issues.
We all know that what we work with are the reads from a sequencing run. On an Illumina platform, we expect to have reads of sizes between 50 nt and 300 nt. Sometimes we have a single fastq file storing these reads from a single-end (SE) run. Other times, we have two, paired fastq files to work with from a paired-end (PE) run. But what is being sequenced? It is actually a piece of DNA with two adapter sequences attached, one at each end. These are used during the sequencing reaction, and when multiple libraries are being run together, they allow for each set of reads to be assigned to the correct library of origin. Below is an outline of what a fragment, with an insert of DNA to be sequenced looks like (in a PE reaction).
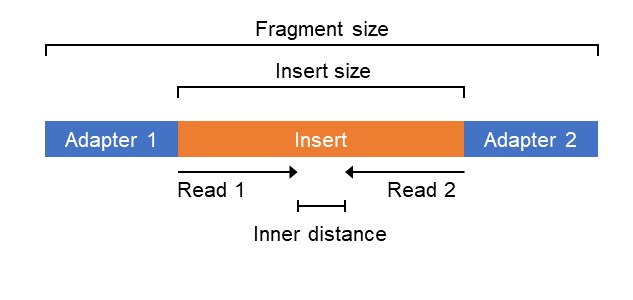
If only a SE run is performed, then only one of the adapters will be used to sequece. If PE, then both adapters are used. With an SE run, you are getting one read per fragment, in PE you are getting two reads per fragment. Why bother getting two reads per fragment? Well it can give you additionaly information, depending on what sort of sequencing you are performing. It can increase the accuracy of mapping the two reads to a reference genome. Or it can help assemble a genome by joining two contigs of a genome together by placing each of those two reads, potentially with a long "inner distance", onto two unconnected contigs. Contigs here are parts of a partially assembled genome.
The insert size is the size of the piece of DNA of interest, without the adapters. This will vary depending on your library prep protocol, which will generate a distabution of inserts of different lengths. Then adapters are added to create the fragment size. If you have an insert of 350 bp, but you perform 150 nt PE sequencing, then you will sequence most of your insert, but there will be an inner distace of 50 bp that is unsequenced. If you have a smaller insert size, let's say 250 bp, but with 150 nt PE reads, then there will be no inner size (see below).

This is not a bad thing, but in a way, you are sequencing the same position twice, which could be seen as a waste of money. Of course, with a distribution of sizes within your library, you would expect this to happen for some fragments, but you might want to aim to make a large enough library that this is not the norm ie that the median insert size of a library is larger than the read length (times two for PE sequencing).
One source of confusion is insert vs fragment. Another is read vs fragment. Especially with the RNA-seq units of measurements RPKM and FPKM. The first stands for Reads Per Kilobase of transcript, per Million mapped reads and the second for Fragments Per Kilobase of transcript, per Million mapped reads. However, there is no effective difference between these units. The term RPKM came about before PE sequencing was around so no one thought to future-proof the term for when PE became popular and we needed to re-centre focus on the fragment. In both cases, one is measuring the relative expression of RNA molecules by the relative fraction of the fragments you are measuring - the only difference is that in SE you only have a single read to help you map/reconstruct that transcript, in PE you have two reads per fragment. So you cannot do any conversion like divide by two, to change between them, they are measuring the same thing.
So this means, if you wanted a large random sampling in your RNA-seq, getting 30 million SE reads might in a way be better than 30 million PE reads. This is because 30 million SE reads is 30 million indepedent fragments, while 30 million PE reads are actually only 15 million fragments, half as many. While each PE read has more mapping confidence or transcript assembly, it probably isn't as valuable as doubling the number of fragments that you get information from.
Finally, if your insert size is smaller than your read length, then you will be reading into the adapter sequence at the other end of the fragment. This means that you will have sequence in your read not matching your sample's genome/transcriptome. Ideally, you should be trimming your reads for adapter sequences, using one of many different tools to do so.

So in summary, an insert is the of DNA of interest you add adapter sequences to. A fragment is the insert plus adapters. A read is the sequenced part of a fragment, usually the insert, but can also sequence parts of the adapters as well. What you are sequencing is the fragment, in either SE or PE sequencing, the only difference is the number of reads per fragment.
Further reading:
http://www.ecseq.com/support/ngs/trimming-adapter-sequences-is-it-necessary
https://www.biostars.org/p/106291/
http://thegenomefactory.blogspot.com.au/2013/08/paired-end-read-confusion-library.html